Benchmarking the Frontier of Drug Discovery: Domain Harnesses Are Key
A biological evaluation of whether a method can surface therapeutically relevant targets from disease data — not merely perform a standard analysis task.
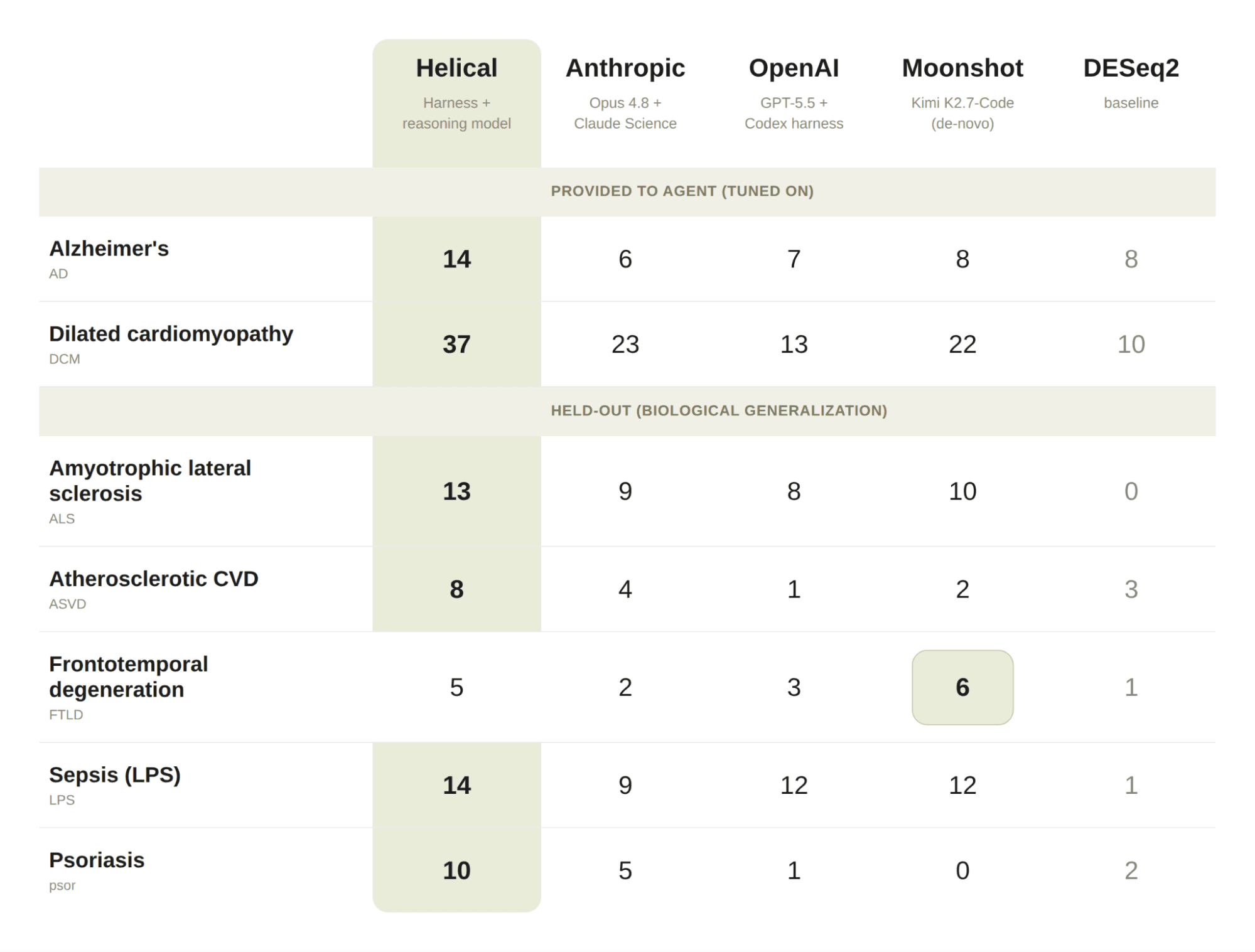
Recovery@100 of therapeutically relevant targets, by indication. Known targets recovered in the top 100 (Open Targets top 5%, shared protein-coding universe, see Benchmark design section). Helical denotes our expert-designed biological harness paired with a reasoning model. Higher is better. Best per row in bold.
1. Motivation
Target identification is one of the most valuable and least forgiving steps in drug discovery: the choice of target shapes every decision downstream. Longitudinal attrition analyses consistently name target selection among the leading technical determinants of program failure [1–3]. Computational target identification is not a new problem: bioinformatic and multi-omic approaches have been applied here for over a decade [4]. What remains largely unaddressed is the evaluation of AI reasoning agents in this context. Existing public benchmarks, including Biomni [5], LAB-Bench [6], BixBench [7], and OpenAI's GeneBench-Pro [8], focus on analytical competence: selecting the right tool, executing a pipeline, or answering a well-defined question about a dataset. These are meaningful checks on an agent's ability to carry out analysis, but they do not establish whether its output constitutes therapeutically meaningful biology, which we define as evidence mechanistically linked to disease, corroborated across independent sources, and specific enough to inform an actual target-selection decision. Here, we propose a framework to evaluate how agents create end-to-end workflows, combining multiple analytical steps, tools, and biological assumptions, to generate this kind of value in practice.
Building benchmarks for this kind of evaluation is difficult. Ground truth is scarce: whether a given target choice was correct is often not established for years, if at all, and attribution of failure to a single cause is rarely possible even in retrospect. Compounding this, the data available for benchmark construction frequently overlaps with data used in method development or validation, introducing a risk of leakage that can inflate apparent performance independent of genuine discovery utility. Given these constraints, we begin with a narrower scenario: evaluating a method's ability to retrieve supporting evidence from transcriptomics, one of the key modalities used in target-level decision making. This allows us to validate platform-constituent workflows in isolation, under controlled conditions, as a first step toward establishing genuine value contribution. Naturally target decisions are not made on transcriptomic evidence alone; this scope reflects a deliberate starting point rather than a complete evaluation, and we intend to extend the benchmark to additional modalities in future work.
Our proposed benchmark therefore asks a practical question: given disease data, can existing agents create reproducible and generalisable workflows for discovery of gene-disease associations supporting target prioritisation? By focusing on workflows we are simulating unbiased discovery from the data rather than retrospective explanation of what is already known. This is, first and foremost, a biological evaluation, and it is the bar we hold our own virtual AI lab to when we run computational experiments on a partner's disease data.
Disclosure. Helical developed this benchmark and is one of the evaluated methods. Conditions are matched across methods, the design is described in sufficient detail to be reproduced and scrutinized, and results are reported for every method, including indications on which the Helical method underperforms.
2. Benchmark design
What task? Given single-cell transcriptomic data for a disease, a method produces a ranked list of candidate protein-coding genes as potential therapeutic targets. We evaluate seven indications spanning several disease areas:
- neurodegeneration, that is Alzheimer's disease (AD) [9], amyotrophic lateral sclerosis (ALS) [10], and frontotemporal lobar degeneration (FTLD) [10];
- cardiovascular disease, that is atherosclerotic cardiovascular disease (ASVD) [11] and dilated cardiomyopathy (DCM) [12];
- inflammatory and immune-mediated disease, that is sepsis, modeled by stimulating patient immune cells via lipopolysaccharide ex vivo [13], and psoriasis (PSOR) [14].
Evaluating across diverse indications is essential: a discovery method should generalise across disease biology rather than overfitting conclusions to a single disease area and potential biases inside models and data.
Why do we score the workflow? Ask a model to name targets for a disease and it can mostly answer from memory: the known targets are in the training data, public databases or literature, so a capable model will list them whether or not it has learned anything from the omics data. Grading that written list measures recall of the literature, not discovery, and a method can score well on it without doing any real biology. To avoid leakage, we reframe the task for each method as writing an indication-agnostic workflow in code that produces a ranked candidate list using only single-cell data as input. This ensures that solutions are data-driven and do not exploit prior knowledge of the indications.
How do we score workflows' outputs? For each indication, we define the positive ground-truth set as the genes whose Open Targets indication-specific association score falls within the top 5% of protein-coding genes for that indication. We report recovery@100: the number of these ground-truth targets that appear within a method's top 100 ranked genes for the same indication. Because association scores are computed per indication and derived externally, largely independent of the single-cell data the method analyzes, recovery tests concordance with independent external evidence rather than internal consistency within the dataset. To avoid circularity, we remove the expression-based (RNA) component from the association score; without this, a method could be credited simply for recovering expression signals that are also embedded in the label. Removing it means recovery reflects independent lines of therapeutic evidence, such as human genetics and known drug-target associations, rather than the expression data the method itself is built on.
3. Experimental design
Biological generalization (development and held-out split). Methods and their thresholds are developed on two indications (AD and DCM). The remaining five (ALS, FTLD, ASVD, Sepsis, PSOR) are held out and used only for evaluation. Because the held-out set spans disease areas the method was not tuned on, performance there tests biological generalization: whether an approach transfers to new disease biology. That is the property that matters for discovery, and it is our primary result. Data is provided raw without any pre-processing.
Prompt design. All prompts are publicly available via GitHub. They were designed to encourage the agents to explore a diverse solution space and not simply default to differential expression analysis, as they appeared to without further guidance.
Matched conditions. All methods are evaluated on the same indications, with the same evaluation procedure and the same protein-coding gene universe as the candidate set.
Reasoning/coding agents. We test the latest models and harnesses available to us, namely:
- Claude Science + Opus 4.8 (Fable 5 unavailable)
- OpenAI Codex + GPT 5.5
- Kimi K2.7-Code
Each of the models is also paired with our own internal Helical harness.
4. Results
Per-indication values are shown in the figure at the top of this page; the main biological findings are summarized below.
Finding 1: Helical's expert-designed harness outperforms general-purpose and domain-specific models across most indications. With our harness — a combination of curated biological models, specialized tools, and scientific reasoning — the agent consistently recovers more therapeutically relevant targets than general-purpose models (Opus 4.8, GPT-5.5, Kimi K2.7-Code) or biology-specific systems (Claude Science) on held-out indications. (Fig. 1)
Finding 2: Individual models can be elevated to the frontier system with the right harness and context in place. The expert-designed harness improves performance across generalist models in the majority of indications tested (e.g., ALS, Sepsis, Psoriasis). (Fig. 2) This suggests that curated biological models, specialized tools, and scientific expertise matter more than the choice of reasoning model alone.
Finding 3: Open models can match the frontier for target discovery. Open models such as Kimi K2.7-Code produce scientifically sophisticated approaches, achieving competitive performance on selected indications (e.g., FTLD). (Fig. 1)
Overall: the system matters more than the model. The expert-designed harness delivers better results than strong general-purpose models: higher target recovery across indications. The key advantage is therefore not any individual general-purpose model, but the system around it.
Generally, the workflows the reasoning agents built without Helical's harness would look familiar to any single-cell analyst. Most began with pseudobulk aggregation, summing each donor's cells before comparing disease to control, which is the field's standard guard against treating cells from one person as independent samples. From there the solutions fanned out across the usual toolkit: differential expression, shifts in cell-type composition, and co-expression networks that flag genes whose wiring to their neighbours changes in disease, an idea with a long lineage in weighted co-expression analysis (WGCNA; Langfelder and Horvath 2008). Pretrained foundation models such as Geneformer and scGPT were tried too, but the agents mostly set them aside once they failed to beat the simpler methods, a caution that matches recent benchmarks showing basic approaches often match large models out of the box. The overall impression is of tools reaching for well-established, defensible practice.

The Helical harness across reasoning models (Recovery@100 by indication). Our expert harness paired with different base models (Opus 4.8, GPT-5.5, Kimi K2.7-Code). The dashed line marks the best general-purpose system for each indication.
5. Limitations
Principal limitations with the published benchmark:
- Test-time compute. Reasoning models can improve their solutions with greater compute budgets where the plateau has not yet been explored, which makes it hard to judge the absolute capability of different reasoning systems under unlimited compute constraints.
- Single modality. The benchmark covers target identification from one data modality and evaluates a single step, not the whole of discovery.
- Proxy for therapeutic truth. Recovery against Open Targets is a proxy, not ground truth. We are exploring avenues to add more depth to our evaluation to paint a clearer picture of where these systems stand.
- Fixed workflow. Constraining methods to a fixed workflow makes results comparable across indications, but suppresses some adaptive behavior that helps in practice. Reported differences should be read within this constraint.
- Coverage. Seven indications across three disease areas is a meaningful but limited slice of biology; we will extend the benchmark to further indications and modalities.
What the metric does not capture: recovery is scored on the ranked candidates alone. In deployment, Helical does not hand back a black-box ranking: every candidate is wrapped in biological evidence, including mechanism, pathways, CRISPR hits, and Open Targets support, so a scientist can interrogate and defend it in a decision committee. That evidence in the loop is what turns a recovered target into a decision package. It is not measured here, which is why recovery is best read as a floor on value rather than the whole of it.
About Helical
Helical is the virtual AI lab for pharma. We are the application layer that orchestrates the world's best bio foundation models, aligned to your biology, so pharma and biotech teams can run scalable, reproducible in-silico experiments across target discovery, biomarker development, patient stratification, and RNA therapy design.
To learn more or schedule an intro call, book a time here.
References
- Cook, D. et al. Lessons learned from the fate of AstraZeneca's drug pipeline: a five-dimensional framework. Nat. Rev. Drug Discov. 13, 419–431 (2014).
- Paul, S. M. et al. How to improve R&D productivity: the pharmaceutical industry's grand challenge. Nat. Rev. Drug Discov. 9, 203–214 (2010).
- Morgan, P. et al. Impact of a five-dimensional framework on R&D productivity at AstraZeneca. Nat. Rev. Drug Discov. 17, 167–181 (2018).
- Katsila, T., Spyroulias, G. A., Patrinos, G. P. & Matsoukas, M.-T. Computational approaches in target identification and drug discovery. Comput. Struct. Biotechnol. J. 14, 177–184 (2016).
- Huang, K. et al. Biomni: A General-Purpose Biomedical AI Agent. Bioinformatics (2025).
- Laurent, J. M. et al. LAB-bench: Measuring capabilities of language models for biology research. arXiv [cs.AI] (2024) doi:10.48550/arXiv.2407.10362.
- Mitchener, L. et al. BixBench: A comprehensive benchmark for LLM-based agents in computational biology. arXiv [q-bio.QM] (2025) doi:10.48550/arXiv.2503.00096.
- Li, J. & Ho, A. GeneBench-Pro: Evaluating multistage statistical reasoning in genomics, quantitative biology, and translational biomedicine. bioRxiv (2026) doi:10.64898/2026.06.29.735386.
- Gabitto, M. I. & Travaglini, K. J. Data resource: Seattle Alzheimer's Disease Brain Cell Atlas (SEA-AD), Allen Institute for Brain Science. Nat. Neurosci 27, 2366–2383 (2024).
- Pineda, S. S. et al. Single-cell dissection of the human motor and prefrontal cortices in ALS and FTLD. Cell 187, 1971–1989.e16 (2024).
- Traeuble, K. et al. Integrated single-cell atlas of human atherosclerotic plaques. Nat. Commun. 16, 8255 (2025).
- Koenig, A. L. et al. Single-cell transcriptomics reveals cell-type-specific diversification in human heart failure. Nat. Cardiovasc. Res. 1, 263–280 (2022).
- Lawlor, N. et al. Single cell analysis of blood mononuclear cells stimulated through either LPS or anti-CD3 and anti-CD28. Front. Immunol. 12, 636720 (2021).
- Reynolds, G. et al. Developmental cell programs are co-opted in inflammatory skin disease. Science 371, eaba6500 (2021).
- Buniello, A. et al. Open Targets Platform: facilitating therapeutic hypotheses building in drug discovery. Nucleic Acids Res. 53, D1467–D1475 (2025).