Recovering Known Targets at Scale
Across 11+ indications in neurology, cardiology, oncology, and immunology, our Virtual Lab recovers known disease-relevant targets at up to a 43% hit rate (i.e. 43 of the top-100 prioritized targets are known with prior biological or experimental support). It outperforms both differential expression (DESeq2) and random baselines on every indication tested, across independent pharma collaborations.
Platform-level consistency across therapeutic areas is the real test. A framework can look compelling on a hand-picked indication. The question is whether it continues to surface disease-relevant biology when applied across independent diseases. That cross-context performance is what gives confidence not just in the known targets recovered (our “calibration test”), but in the novel targets and pathways prioritized beyond the known set. This doesn't prove every novel prediction is correct, but systematic enrichment across diverse indications is the prerequisite for taking novel predictions seriously. Several of those novel candidates are now advancing into wet-lab follow-up with our pharma partners.

From Association to Restoration: What Target ID Has Been Missing
So how does this work, and why does it matter?
Target identification is one of the highest-leverage decisions in drug discovery. The right target can shape the trajectory of an entire program. The wrong one can cost years of work and substantial experimental investment.
Most approaches today remain largely observational. They rank targets based on what is associated with disease: differential expression, genetic association, static pathway evidence. All of these signals can tell you what is altered in disease, but none directly tell you what is most likely to restore a healthy phenotype. Bio foundation models (BioFMs) have since brought far richer representations of disease biology, but most workflows still apply them to the same question.
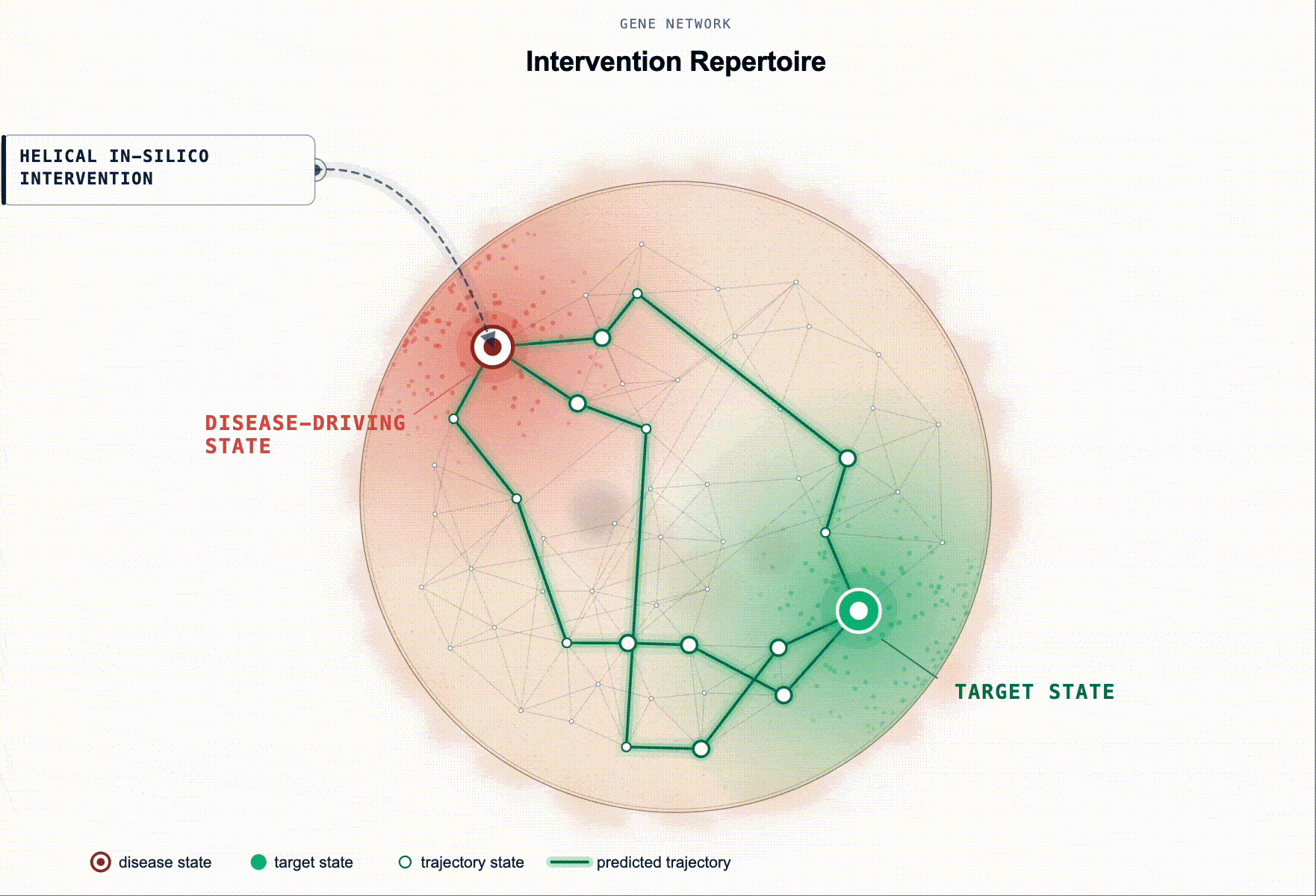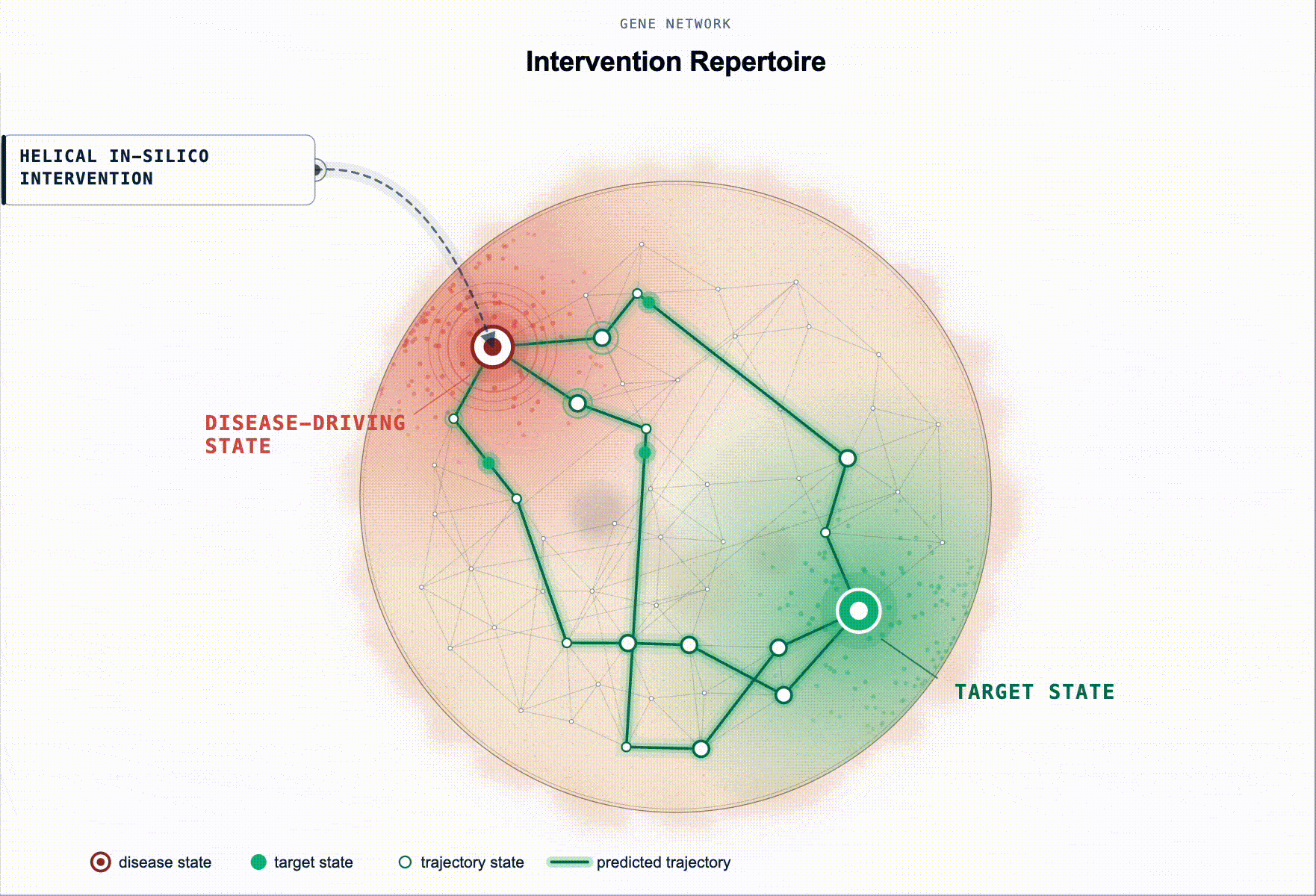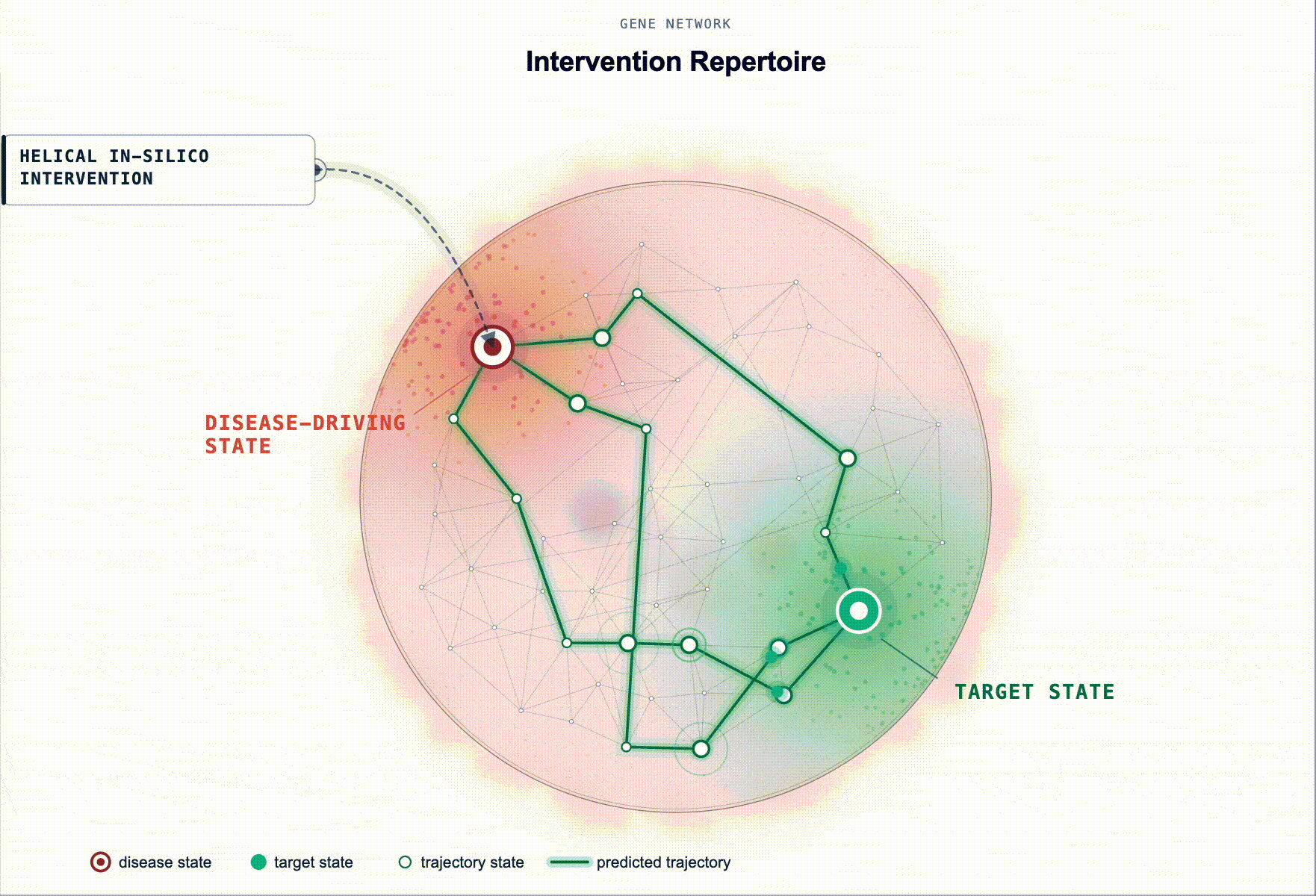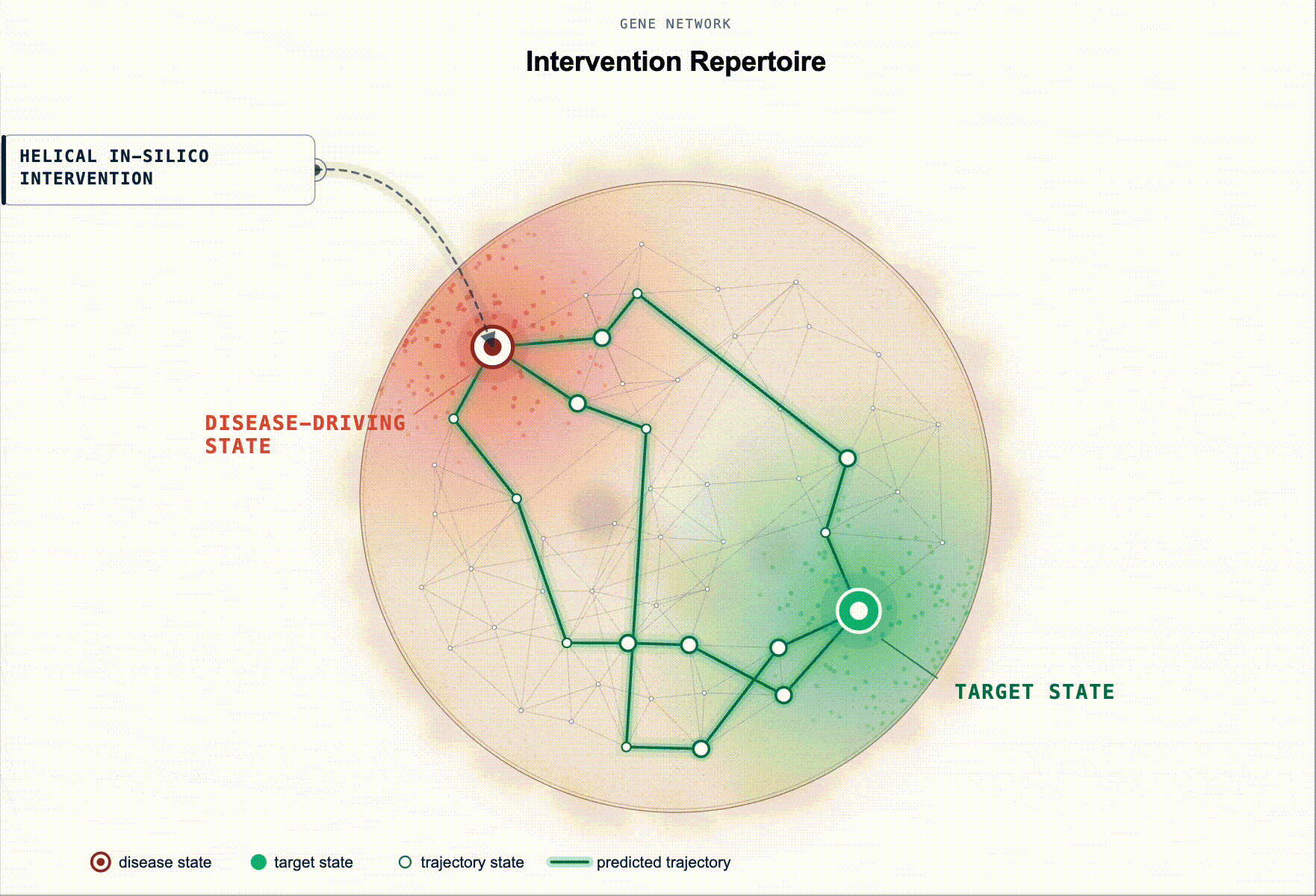
What has been missing is not more association evidence but an executable disease model. Something a scientist can intervene on in-silico and ask: “if I knock these genes down in this cell state, does the system restore?”
That is the question drug discovery must answer. Association evidence is essential, but insufficient to justify intervention.
Rather than ranking by association, our framework prioritises targets by their predicted capacity to restore a healthy state. Starting from a disease-tailored BioFM, we systematically perturb genes in-silico and rank them according to whether the perturbation shifts the modelled disease-driving cell state back toward health. For multi-cellular disease, this principle extends to reshaping the disease-driving communication program between cell populations.
This turns target ID from association ranking into in-silico hypothesis testing: which perturbations are most likely to produce a meaningful restorative effect, in which cell types, and under which disease contexts.

Biology First: The Right Model and the Right Workflow for the Disease (Model Factory)
Making this work across indications requires more than a good model. It requires a system that adapts to each disease context.
A core lesson from building the Virtual Lab is that there is rarely a single universal winner in biology. The most hyped model is often not the one that performs best for a specific disease setting. And even with the right model, the same workflow cannot simply be applied everywhere.
That is why we built a Model Factory: a systematic framework to tailor, benchmark, and select models against disease biology rather than defaulting to whichever foundation model is currently most talked about. Models alone do not make discoveries. The system built around them — how they are selected, adapted, and matched to the biology — is what makes their outputs meaningful. The Model Factory is that system.
Just as importantly, the downstream workflow adapts to the mechanism of disease. We have different targeting angles depending on the pathology and the biology driving it. Figure 3 illustrates this process: we first benchmark candidate foundation models against disease-relevant evaluation axes, select the strongest performer, and then systematically evaluate training adaptations of that model to identify the configuration best suited to the biology at hand.
In some settings, the key problem is cell-intrinsic disease biology, where in-silico perturbation of cellular state is the right abstraction. Here, we ask which perturbations restore dysfunctional cells toward healthier reference states.
Where pathology is driven by cell–cell interaction (CCI), single-cell state alone cannot capture the relevant biology. In inflammatory conditions like psoriasis, rheumatoid arthritis, and IBD, for instance, Th17 cells are central drivers, but their disease role plays out through communication with other immune and tissue-resident populations. Ranking targets within one cell type would miss the intercellular program that actually sustains disease. That is why we use CCI-adapted models and workflows that explicitly model intercellular signalling programs and ask how perturbations reshape disease-driving interactions across cell types.
Other settings call for different framing entirely: isoform-level resolution where splice variants drive distinct disease biology that gene-level analysis would conflate, spatial context where tissue architecture shapes pathology in ways dissociation-based data cannot preserve, or perturbation-experiment priors where existing screens and genetic studies provide direct causal evidence that should anchor in-silico perturbation-effect predictions.
Biology has to come first. The model, the benchmark, and the workflow all need to be fit for purpose. That is how target ID generalises: not by forcing every disease through the same pipeline, but by matching the model and workflow to the biology actually driving pathology.


What Helical's Virtual Lab Unlocks
Target ID is one instantiation of a more general framework. The same in-silico lab, the same Model Factory, the same expertly crafted toolkits and workflow rubrics, unlocks a broader set of capabilities:
- Target identification — systematic in-silico perturbation and validation to identify targets that reshape disease-driving cell states and intercellular programs.
- Cell-state discovery — identifying disease-driving cell states that should be modeled, perturbed, or validated.
- Patient stratification — identifying patient subgroups that share disease-driving biology and are therefore more likely to respond to a given intervention.
- Genomic integration — linking sequence variation to predicted effects on isoforms, regulation, and disease biology.
- Multi-omics modeling — integrating the most relevant molecular and tissue-level modalities — single-cell, bulk, spatial, genomic, proteomic, and clinical data — into a shared disease model.
- Therapeutic design — supporting antisense oligonucleotide (ASO) design and modeling before experimental screening.
These workflows define how disease state is represented, how perturbations are simulated, how restorative effects are quantified, and how outputs are turned into prioritisation decisions. The value is not just in representation learning. It is in translating model outputs into decision-ready hypotheses that are mechanistically grounded, disease-contextualised, and ready for experimental follow-up. This is where most of the field still falls short.

What's Next
A scientific manuscript with multiple pharma partners is forthcoming, detailing the framework, benchmarks, and case studies. We are grateful for these collaborations, and more are coming.
Wet-lab validation of platform-derived candidates is underway. We are also expanding the in-silico lab across additional therapeutic areas and deeper multi-omic integration. We look forward to sharing more as partner programs and validation studies progress.
If you are working on translational target ID, or any of the capabilities above, and this resonates, get in touch.